Energie
potencjalne w oddziaływaniach międzymolekularnych
|
Typ
oddziaływań |
Zależność
potencjału U od odległości międzymolekularnej R |
Typowa
energia kJ/mol |
Uwagi |
|
Jon-jon (Q1, Q2) (1/R) |
-
Q1Q2/(4πε0R) |
250 |
Tylko
między jonami |
|
Jon-dipol
(Q, μ) (1/R2, 1/R4) |
- Qμ cos θ/(4πε0R2) - Q2μ2/6 (4πε0)2kTR4 |
15 |
Oś dipola tworzy kąt θ z osią łączącą dipol z ładunkiem Dipol
rotujący swobodnie w temperaturze T |
|
Jon - molekuła niepolarna (Q, 0) (1/R4) |
-
Q2α/2 (4πε0)2R4 |
|
Między
jonem a niepolarną molekułą o polaryzowalności α |
|
Dipol-dipol (μ1, μ2) (1/R3, 1/R6) |
- μ1μ2[2 cos θ1 cos
θ2 - sin θ1 sin θ2 cos n]/4πε0R3 - μ12
μ22/3 (4πε0)2kTR6 |
2
0.3 |
Między trwałymi dipolami w ciele
stałym tworzącymi kąty θ1 i θ2 z osią łączącą i
skręconymi o kąt φ. Między trwałymi dipolami
rotującymi swobodnie w płynach (energia
Keesoma) |
|
Dipol-dipol indukowany (μ, 0) (1/R6) |
- μ2α(1+ 3cos2θ)/2(4πε0)2R6 - μ2α/(4πε0)2R6 |
|
Między molekułą niepolarną i
trwałym nieruchomym dipolem. Oś dipola tworzy kąt θ z osią łączącą dipol z molekułą niepolarną. Między molekułą niepolarną i
trwałym dipolem rotującym swobodnie. (energia Debye=a) |
|
Dyspersyjne Londona (1/R6) |
-
3 hvα2/4 (4πε0)2R6 |
2 |
Między
wszystkimi typami molekuł. hv jest energią jonizacji. |
Energia wiązania wodorowego A-H..B
ma typową wartość 20 kJ/mol. Wiązanie występuje przy kontakcie molekuł dla A, B
= N, O lub F.
ENERGIE POTENCJALNE W PROSTYCH ODDZIAŁYWANIACH MIĘDZYMOLEKULARNYCH
Elektrostatyczne
|
Jon - jon
Jon - dipol
Dipol - dipol
|
|
|||||||||
|
Dyspersyjne
|
Dipol
indukowany - dipol indukowany
|
|
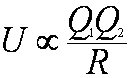
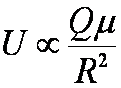
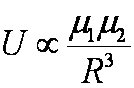
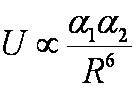
Zależność
między siłą oddziaływania F a
energią potencjalną U:

Potencjały modelowe w oddziaływaniach
molekuł w ciekłych kryształach
Ogólne kwantowomechaniczne wyrażenie na oddziaływania międzymolekularne jest krańcowo skomplikowane i w zasadzie nieużyteczne w praktycznych wyliczeniach, w tym także w symulacjach komputerowych. Jednak kluczem prostoty w tym wyrażeniu jest zwykła energia potencjalna Coulomba oddziaływań między dwoma ładunkami punktowymi
![]()
W wielu przypadkach, zwłaszcza dla molekuł prętopodobnych, można otrzymać uproszczone wyrażenia. Kilka takich uproszczonych potencjałów oddziaływania stosuje się w symulacjach procesów w ciekłych kryształach. Należą tutaj głównie potencjały Luckhursta - Romano, i Gaya - Berne’a oparte na bardzo wygodnym i popularnym czysto przyciągająco-odpychającym potencjale Lennard-Jonesa gdzie U jest potencjałem oddziaływania między dwiema cząstkami, których środki są odległe o R. (LenJones3D.wpg.gif)
:
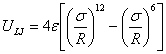
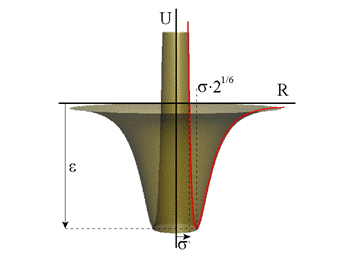
Tutaj ε jest głębokością minimum potencjału, a σ - położeniem zera potencjału. Wykładnik 6 jest typowy dla oddziaływań dyspersyjnych, natomiast wykładnik 12 nie ma znaczenia fizycznego ale jest wybrany dla wygody w wyliczeniach i obrazuje silne oddziaływania odpychające. W tym potencjale punkt o wartości minimalnej Umin = - ε znajduje się w odległości R0 = σ·21/6. Parametr σ daje zgrubną ocenę rozmiaru molekuły. Z drugiej strony, czysty potencjał kątowy, oparty na wielomianie Legendre’a drugiego rzędu, P2 (x) = (3x2 - 1)/2, został zaproponowany przez Lebwohla i Lashera:
![]()
Tutaj ![]() jest wektorem
jednostkowym określającym orientację przestrzenną osi symetrii molekuły.
Później dołączono do tego potencjału człon uwzględniający przyciąganie między
molekułami:
jest wektorem
jednostkowym określającym orientację przestrzenną osi symetrii molekuły.
Później dołączono do tego potencjału człon uwzględniający przyciąganie między
molekułami:
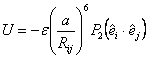
gdzie a jest odległością między najbliższymi sąsiadami i ponownie P2 (x) = (3x2 - 1)/2.
W ostatnich latach w symulacjach ciekłych kryształów używa się głównie dwóch potencjałów modelowych: Luckhursta i Romano oraz Gaya i Berne’a. Oba te potencjały można traktować jako kombinację potencjału Lennard-Jonesa z odpowiednimi członami opisującymi oddziaływania orientacyjne. Potencjał Luckhursta i Romano ma postać:
![]()
za najlepszą wartość parametru λ wybrano λ = 0.15 . Obliczenia przeprowadzano dla dwóch próbnych funkcji F(R): F1(R) = 4ε (σ/R)6 oraz F2(R) = 4ε[ (σ/R)12 + (σ/R)6 ].
Potencjał Berne’a i Gay’a ma postać:
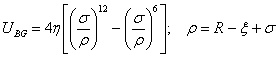
gdzie
![]()
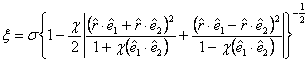
![]() jest wektorem jednostkowym określającym kierunek
między molekułami 1 i 2, zaś
jest wektorem jednostkowym określającym kierunek
między molekułami 1 i 2, zaś
χ (0 < χ < 1) opisuje anizotropię molekuł. Wartość χ = 0 oznacza molekuły kuliste i wtedy potencjał Berne’a i Gay’a przechodzi w potencjał Lennard-Jonesa natomiast χ = 1 oznaczałaby molekuły o kształcie nieskończenie cienkich prętów. (Berne-Gay.wpg.gif)
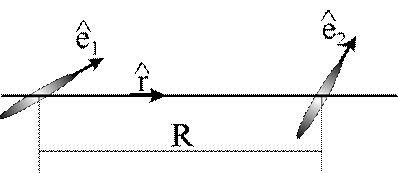
Oddziaływania między określoną molekułą a wszystkimi pozostałymi w układzie mogą być wyliczane przy założeniu addytywności oddziaływań dwucząstkowych. Jednak, aby uprościć wyliczenia dla potencjałów krótkozasięgowych, należy używać potencjałów obcinanych i ograniczyć sumowanie do określonego sferycznego obszaru obcięcia.
WIĄZANIA WODOROWE
Wiązanie wodorowe polega na zdolności atomu wodoru H do wiązania dwóch bardzo elektroujemnych atomów N, O lub F. Wiązanie wodorowe - jest częściowo (ok. 90%) elektrostatyczne, a częściowo (ok.10%) kowalencyjne. Wiązania wodorowe są ukierunkowane i w przypadku wody wzajemna konfiguracja molekuł jest przez te wiązania ściśle określona.
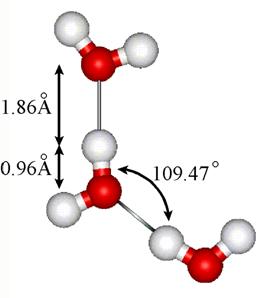
Struktury przestrzenne, powstające w wodzie w wyniku wiązań wodorowych, są tworzone bardzo dynamicznie ponieważ średni czas trwania wiązania wodorowego w wodzie w temperaturze pokojowej wynosi 3×10-12 s (3 ps). Energia wiązania wodorowego wynosi ok. 4 - 25 kJ/mol. Wiązania wodorowe mają wpływ na:
- obniżenie ciśnienia pary
- podwyższenie temperatury wrzenia
- podwyższenie lepkości
- niską gęstość lodu
- hydratację przez wodę
- rozpuszczalność w wodzie
- sztywność struktury
- organizację konformacyjną (kształt makromolekuł biologicznych)
Najważniejsze zjawiska wywołane przez wiązanie wodorowe:
* Tworzenie się otwartych struktur wody i lodu (lód pływa po wodzie, co jest wyjątkowym w przyrodzie zjawiskiem pływania kryształu na cieczy powstałej z jego stopienia)
* Formowanie się podwójnej helisy DNA
* Formowanie się helisy α białek.