Zebranie natężeń refleksów jest ostatnim etapem doświadczalnym. Dalsze postępowanie
polega wyłącznie na obliczeniach komputerowych.
Sposób pomiaru natężeń refleksów zależy od typu dyfraktometru, ale zasada
pomiaru jest zawsze taka sama. Refleksy uważa się za punktowe, lecz w rzeczywistości
mają one skończone wymiary. Z tego względu do pomiaru ich natężeń wykonuje
się tzw. skanowanie (przemiatanie). Stosowane są przy tym dwie techniki: ω-skanowanie
i ω/2θ-skanowanie. W technice ω
przy teoretycznym kącie 2θ dla mierzonego refleksu, detektor
(licznik) pozostaje nieruchomy, natomiast kryształ obraca się o kąt ω
(1—2°). W technice ω/2θ porusza się zarówno monokryształ,
jak i detektor, przy czym obrotowi kryształu o kąt Δω
towarzyszy przesuw detektora po kole 2θ z prędkością kołową
dwukrotnie większą od prędkości obrotu kryształu (Δ2θ = 2Δω).
Technika ω jest szybsza od techniki ω/2θ,
wymaga jednak zastosowania monochromatorow grafitowych w celu uzyskania dobrze
zmonochromatyzo-wanego promieniowania. Przy stosowaniu filtrów polecana jest
technika ω/2θ.
Ze względów konstrukcyjnych goniostat Eulera umożliwia pomiar natężeń dla
kątów 0≤2θ≤150°. W praktyce cały zakres wykorzystuje
się tylko niekiedy przy zastosowaniu promieniowania CuKα, dla
którego przy 2θ=150° sinθ/λ ma wartość
ok. 0,63Å-1. Dla promieniowania MoKα podobną
wartość sinθ/λ osiąga się już dla 2θ≈50°.
Dla większych wartości sinθ/λ natężenia zmierzonych
refleksów są zbyt małe, szczególnie w przypadku związków organicznych zbudowanych
głównie z lekkich atomów. Całkowita liczba refleksów W możliwych
do zarejestrowania zależy od wartości θmax, długości
fali promieniowania rentgenowskiego i objętości komórki elementarnej zgodnie
ze wzorem:
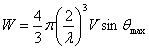
Refleksy słabe o natężeniu I mniejszym od oszacowanego błędu pomiaru
natężeń σ(I) nie są wykorzystywane w obliczaniu struktury. W dobrym
pomiarze liczba refleksów słabych nie powinna przekraczać 10—30% wszystkich
zmierzonych refleksów. Wielu współczesnych badaczy do badań struktury wykorzystuje
tylko refleksy spełniające zależność I≥nσ(I), gdzie n
wynosi 3—6. Duża liczba odrzuconych refleksów ma odbicie w dokładności wyników
pomiarów. Duży procent słabych refleksów stwarza niebezpieczeństwo niepowodzenia
w analizowaniu struktury. Należy w takim przypadku zmienić warunki pomiaru,
a nawet użyć nowego monokryształu.
Jak już wcześniej podano, liczba mierzalnych refleksów zależy m.in. od długości
użytego promieniowania oraz od objętości wybranej komórki elementarnej. Na
ogół dla rozwiązania struktury przeciętnej wielkości związku organicznego
wystarczy analiza natężeń od kilkuset do kilku tysięcy refleksów.
Promienie rentgenowskie ulegają rozproszeniu na wszystkich atomach (ściślej
na ich elektronach) wchodzących w skład badanego związku. Różne rodzaje atomów
mają różną zdolność rozpraszania zależną od całkowitej liczby ich elektronów.
Im atom cięższy, tym więcej ma elektronów, a więc tym intensywniejszy refleks.
Wiązki promieni ugiętych na płaszczyznach atomowych różnych atomów interferują
ze sobą, prowadząc do zmian natężeń (amplitud) wiązek ugiętych. Fakt, że natężenia
wiązek ugiętych zależą od różnic w zdolnościach rozpraszających różnych atomów,
umożliwia zlokalizowanie różnych atomów w komórce elementarnej, a tym samym
określenie struktury związku.
Uproszczony opis analizy natężeń refleksów
Ścisła zależność pomiędzy względnymi natężeniami refleksów (promieni ugiętych)
oraz rozkładem gęstości elektronowej w komórce elementarnej
jest bardzo złożona.
Jak już wspomniano, pomiar natężeń wiązek ugiętych ma na celu znalezienie funkcji
rozkładu gęstości elektronowej w komórce elementarnej.
Z teorii dyfrakcji nie wynika wprost zależność pomiędzy natężeniami wiązki ugiętej
(natężeniami refleksów) a gęstością elektronową, a więc i położeniem atomów
w komórce elementarnej. Dla większości refleksów dane doświadczalne nie dostarczają
bowiem ich tzw. fazy (promieniowanie jest falą o amplitudzie zależnej od fazy).
Stosunkowo niedawno wykorzystano fakt, że w każdym dyfraktogramie można znaleźć
pewną liczbę refleksów o fazach ustalonych (zbiór startowy). Wychodząc z tego
zbioru startowego można ustalić fazy pozostałych refleksów, a tym samym rozwiązać
problem fazowy, a więc strukturę cząsteczki. Metody opierające się na takim
sposobie rozwiązania problemu fazowego nazwano metodami bezpośrednimi.
Dawniej stosowano najczęściej metodę pośrednią zwaną
metodą ciężkiego atomu, wykorzystującą względną łatwość ustalenia położenia
tego atomu.
W metodach pośrednich inaczej się postępuje przy wyznaczaniu faz dla struktur
(komórek) centrosymetrycznych i inaczej przy niecentrosymetrycznych. Badanie
struktur niecentrosymetrycznych jest znacznie trudniejsze, częściej następują
niepowodzenia. Wszystkie obliczenia niezbędne do rozwiązania struktury metodami
bezpośrednimi wykonuje się automatycznie przy wykorzystaniu odpowiednich pakietów
programów komputerowych.
|
Rys. 22. Integracja refleksów przez program komputerowy. |
Udokładnianie i wskaźnik rozbieżności
Model struktury otrzymany na podstawie obliczeń wykonanych metodami bezpośrednimi
można wyrazić matematycznie za pomocą szeregu parametrów. Należą do nich w pierwszym
rzędzie współrzędne położeń cięższych atomów w komórce elementarnej. Położenia
lżejszych atomów, a szczególnie atomów wodoru, najczęściej wyznacza się tylko
orientacyjnie, a niekiedy pomija się w wyniku, lokując wstępnie w pozycjach
wynikających ze znanych, przeciętnych długości wiązań wodoru z innymi atomami
oraz wielkości spodziewanych kątów walencyjnych.
Wszystkie parametry położeniowe atomów uzyskane na etapie rozwiązania struktury
metodą bezpośrednią należy poprawić.
Do oceny poprawności modelu struktury stosuje się wartość tzw. wskaźnika rozbieżności
R. Wartość R jest to względna średnia różnica między amplitudą
struktury obliczoną z modelu i wyznaczoną doświadczalnie na podstawie znanej
liczby refleksów. Wielkość R jest bezwymiarowa, czasami podaje się
jej wartość w procentach, na ogół w postaci ułamka dziesiętnego. Wartość R
można traktować jako wskaźnik stopnia odzwierciedlenia przez model rzeczywistej
struktury cząsteczki. Jako wielkość względna jest ona z definicji zawsze mniejsza
od 1. Dla przypadkowej struktury umieszczonej losowo w komórce elementarnej
można również obliczyć współczynnik R — w takim przypadku dość znacznie
różni się on od 1 i zależy tylko od charakteru grupy przestrzennej. Dla grupy
centrosymetrycznej R < 0,828, dla niecentrosyme-rycznej R
< 0,586. Jest więc oczywiste, że wartość R odnosząca się do rozwiązania poprawnego,
nawet bardzo przybliżonego, musi być znacznie mniejsza od odpowiednich wartości
R podanych powyżej. Wartości R obliczone metodami bezpośrednimi są często bardzo
duże (0,1—0,45). Nie dowodzą one poprawności rozwiązań. W celu poprawienia wyniku
(zmniejszenia wartości R) stosuje się udokładnienie
polegające na dopasowywaniu wartości parametrów metodą najmniejszych kwadratów,
z uwzględnieniem m.in. różnej dokładności wyznaczenia amplitud struktury dla
różnych refleksów. Udokładnianie (dopasowanie parametrów) powtarza się wiele
razy, aż do uzyskania zadowalającej wartości współczynnika rozbieżności R.
Stosując do udokładniania metodę najmniejszych kwadratów uzyskuje się również
odchylenia standardowe udokładnionych parametrów.
W dotychczasowych rozważaniach celowo nie uwzględniono drgań atomów w siatce
krystalicznej i wynikających z tego tzw. czynników temperaturowych (izotropowych
i anizotropowych). Położenia atomów wodoru w komórce elementarnej są wyznaczone
w przybliżeniu o rząd wielkości mniej dokładnie niż pozostałych atomów, stąd
wystarczy dla nich uwzględnić jedynie izotropowe czynniki temperaturowe. Parametry
(czynniki) temperaturowe atomów podaje się zwykle w tabelach parametrów pozycyjnych
atomów.
Po uwzględnieniu wszystkich poprawek końcowa wartość współczynnika rozbieżności
R powinna być mniejsza od 0,05. Położenia atomów cięższych w modelu
końcowym o R < 0,05 wyznaczane są zwykle z dokładnością większą
niż 5*10-4 Å, atomów wodoru z dokładnością o rząd mniejszą.
Parametry pozycyjne atomów, długości wiązań, kąty walencyjne i torsyjne
Każda komórka elementarna jest równoległościanem. Jej przedłużone krawędzie
można traktować jako osie układu współrzędnych, a długości krawędzi jako jednostki.
Znając parametry komórki elementarnej (długości krawędzi i kąty między nimi)
i wybierając początek układu w jej narożu, każdy punkt przestrzeni można w tym
układzie współrzędnych zdefiniować jednoznacznie przez podanie trzech bezwymiarowych
parametrów x/a, y/b i z/c (często w literaturze oznaczane
są one po prostu x, y, z). Te bezwymiarowe parametry nazywane są współrzędnymi
ułamkowymi pozycyjnymi atomu traktowanego w sieci krystalicznej jako punkt.
Gdy atom leży wewnątrz komórki elementarnej, wówczas wartości jego współrzędnych
ułamkowych leżą w granicach od 0 do 1. Wartość 0 lub 1 oznacza, iż atom leży
na krawędzi komórki, wartość ujemna lub większa od 1 wskazuje, że atom wychodzi
poza komórkę elementarną. Ze współrzędnych ułamkowych atomów można obliczyć
odległości międzyatomowe, w tym również długości wiązań, kąty walencyjne i torsyjne.
