POTENCJAŁY
TERMODYNAMICZNE
MINIMA
POTENCJAŁÓW W STANACH RÓWNOWAGI
POTENCJAŁY
TERMODYNAMICZNE W PRZEMIANACH FAZOWYCH
GRADIENTY
I ZJAWISKA TRANSPORTU
* Minima potencjałów termodynamicznych w stanach równowagi
* Potencjały termodynamiczne w przemianach fazowych
* Gradienty i zjawiska transportu

Potencjały
termodynamiczne są funkcjami stanu, których zmiana wartości w procesach
równowagowych (odwracalnych) przy ustalonych wartościach określonych par
parametrów stanu ( T i V, T i P, S i P, S i V itd.) jest równa pracy
zredukowanej W*, tzn. całkowitej pracy zmniejszonej o pracę wykonaną na
przezwyciężenia ciśnienia zewnętrznego. Zdolność do przekształcania w pracę
jest powodem, że te funkcje zostały nazwane potencjałami. Istnieje pewna liczba potencjałów
termodynamicznych, które mogą być użyte do opisu zachowania się i stabilności
układów termodynamicznych, zależnie od typu nałożonych ograniczeń w układzie. W
układzie, który jest izolowany od reszty świata, energia wewnętrzna jest
wielkością, która przyjmuje wartość minimalną w stanie równowagi. Jeżeli jednak
połączymy nasz układ termicznie, mechanicznie albo chemicznie ze światem
zewnętrznym, wtedy minimalizacji podlegają inne potencjały termodynamiczne.
Wprowadzimy pięć powszechnie używanych potencjałów termodynamicznych: energia wewnętrzna U, energia swobodna
F, entalpia H, entalpia swobodna G i
potencjał chemiczny :, a także przedyskutujemy warunki w jakich każdy z nich jest
minimalizowany w stanie równowagi. Przedstawione
to zostało na schemacie poniżej dla energii wewnętrznej U, energii swobodnej F,
entalpii H i entalpii swobodnej (potencjału Gibbsa, funkcji Gibbsa) G. Funkcja :, zwana potencjałem chemicznym,
wchodzi w skład potencjałów termodynamicznych w przypadku układów otwartych ze
zmienną liczbą molekuł.
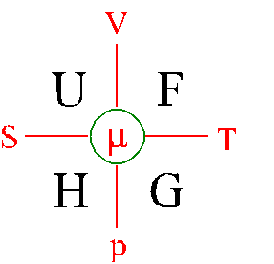
Niezależne S i V: energia wewnętrzna U
Niezależne V i
T: energia swobodna F
Niezależne S i
p: entalpia H
Niezależne T i
p: entalpia swobodna G
1. Niezależne
S i V: stała energia wewnętrzna U
Na
podstawie pierwszej zasady termodynamiki
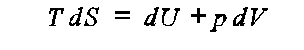
przy S, V = const, mamy dS = 0 oraz dV = 0, zatem
![]()
czyli przy ustalonych S i V energia wewnętrzna U jest stała, U = const.
2. Niezależne
T i V: stała energia swobodna F (energia swobodna Helmholtza).
Na
podstawie pierwszej zasady termodynamiki
![]()
przy T, V = const, mamy dV = 0 i
możemy napisać
![]()
czyli przy ustalonych T i V
wielkość F = U - TS jest również stała
![]()
Funkcja F jest właśnie energią
swobodną. Jej zmniejszenie jest równe pracy wykonanej przez układ
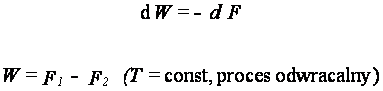
3. Niezależne
S i p: stała entalpia H
Na
podstawie pierwszej zasady termodynamiki
![]()
przy S, p = const, mamy dS = 0 i
możemy napisać
![]()
czyli przy ustalonych S i p
wielkość H = U + pV jest również stała
![]()
Funkcja stanu H = U + pV jest
nazywana entalpią.
4. Niezależne
T i p: stała entalpia swobodna G
Entalpia
swobodna jest nazywana także energią swobodną Gibbsa, potencjałem izotermicznym
i funkcją Gibbsa.
Na
podstawie pierwszej zasady termodynamiki
![]()
przy T, p = const możemy napisać
![]()
czyli przy ustalonych T i p
wielkość G = U + pV - TS jest również stała
![]()
Funkcja stanu G = U + pV - TS jest
nazywana entalpią swobodną.
5. Zmienna liczba cząstek N: potencjał chemiczny μ
Ciśnienie i temperatura nie są wielkościami addytywnymi. W
stanie równowagi ich wartości są takie same w każdej części układu. Takie
wielkości nazywane są intensywnymi.
W układach, w których ilość cząstek jest zmienna,
szczególne miejsce wśród czterech potencjałów termodynamicznych
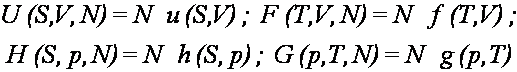
zajmuje
entalpia swobodna G. Przyczyna tej osobliwości polega na tym, że potencjał
właściwy, czyli g(p, T) okazuje się funkcją jedynie zmiennych intensywnych
p i T. Funkcja g(p, T), tj. potencjał termodynamiczny liczony na jedną
molekułę, nazywa się potencjałem chemicznym
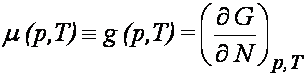
Potencjał chemiczny można także wyrazić jako pochodne
pozostałych potencjałów termodynamicznych względem liczby molekuł N.
Na
podstawie określenia energii wewnętrznej dla układów otwartych
![]()
możemy
napisać
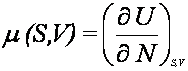
Ponieważ
![]()
oraz
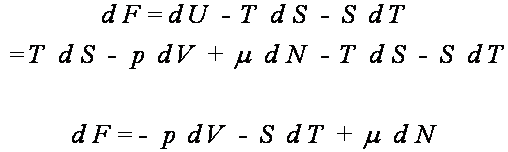
stąd
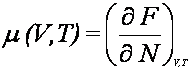
Następnie
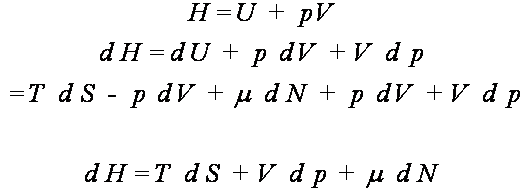
Stąd
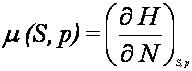
W ten
sam sposób
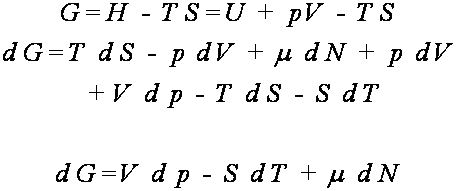
Stąd ponownie
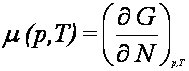

W procesach nieodwracalnych, gdy dQ < TdS, gdy układ
zdąża do stanu równowagi przy ustalonej parze parametrów, to odpowiednie
potencjały termodynamiczne maleją. Łatwo udowodnić, że po osiągnięciu stanu
równowagi potencjały te przyjmują wartości minimalne.
Osiąganie przez potencjały minimum w
stanach równowagowych układu przy ustalonej odpowiedniej parze parametrów stanu
jest najbardziej użyteczną cechą, ze względu np. na symulacje komputerowe.
Proces osiągania przez potencjały wartości minimalnych opisujemy następująco:
We wszystkich przypadkach posługujemy się własnością
entropii
![]()
1. Ustalamy S i V - analizujemy U: biorąc pod uwagę, że dS = 0 oraz dV = 0, a następnie
dzieląc przez różniczkę czasu dt otrzymujemy
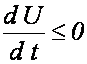
Konkluzja:
proces nieodwracalny przebiegający przy stałych S i V prowadzi do stanu
równowagi, w którym układ ma minimum energii wewnętrznej U.
2. Ustalamy T i V - analizujemy F: biorąc pod uwagę, że dV =
0, a następnie dzieląc przez różniczkę czasu dt otrzymujemy
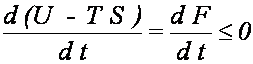
Konkluzja: proces
nieodwracalny przebiegający przy stałych T i V
prowadzi do stanu równowagi, w którym układ ma
minimum energii swobodnej F.
3. Ustalamy S i p - analizujemy H: biorąc pod uwagę, że dS =
0, a następnie dzieląc przez różniczkę czasu dt otrzymujemy
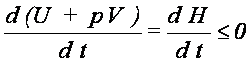
Konkluzja: proces
nieodwracalny przebiegający przy stałych S i p
prowadzi do stanu równowagi, w którym układ ma
minimum entalpii H.
4. Ustalamy p i T - analizujemy G: biorąc pod uwagę, że dS =
0, a następnie dzieląc przez różniczkę czasu dt otrzymujemy
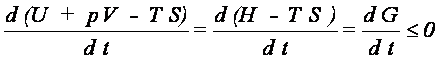
Konkluzja: proces nieodwracalny przebiegający przy stałych p i T prowadzi do stanu równowagi, w którym układ ma minimum entalpii swobodnej G.

Opis
termodynamiczny przemian fazowych I-go i II-go rodzaju zwykle jest oparty o
analizę energii swobodnej Helmholtza (F) lub energii swobodnej Gibbsa (G).
Funkcja Gibbsa G układu pozostaje niezmieniona niezmieniona w każdym procesie,
który przebiega w stałej temperaturze i pod stałym ciśnieniem. (G = U –
TS + pV, zatem dG = dU – TdS – SdT + pdV + Vdp = Vdp – SdT,
ponieważ dU – TdS + pdV = 0). Ponieważ przemiana fazowa zawsze zachodzi
przy określonych wartościach tych dwóch parametrów to funkcja G zachowuje
ciągłość w czasie tej przemiany.
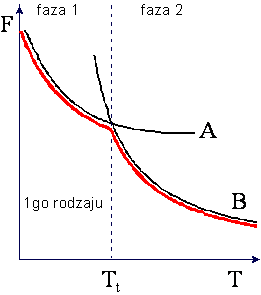
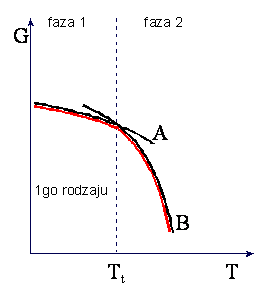
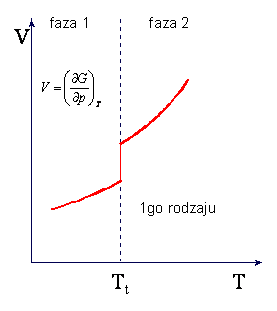
W przemianie fazowej I-go rodzaju
zachodząca wtedy zmiana objętości i istnienie ciepła przemiany powodują, że w punkcie
przemiany nieciągłe są pochodne
![]() oraz
oraz ![]()
ponieważ musi wystąpić zmiana
entropii S wywołana ciepłem przemiany Q/T. Skoki objętości i entropii w
przemianie I-go rodzaju są przedstawione na rysunkach
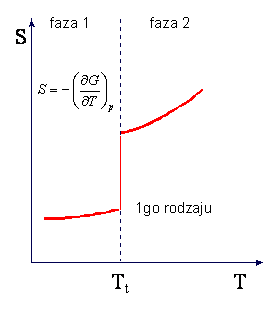
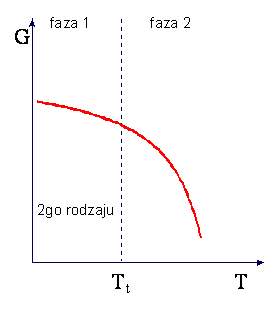
W przemianie fazowej II-go rodzaju nieciągłe są pochodne
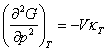 oraz
oraz 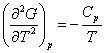
podczas gdy pochodne pierwszego
rzędu, -S i V, są ciągłe. W punkcie przemiany II-go rodzaju występuje zmiana
tylko w nachyleniu krzywych entropii i objętości w funkcji temperatury podczas
gdy nieciągłości występują dla ciepła właściwego i dla izotermicznego modułu
objetościowego (κT = (naprężenie izotermiczne)/(zmiana
objętości)). Charakter zmian entropii i ciepła właściwego w przemianie II-go
rodzaju są przedstawione na rysunkach
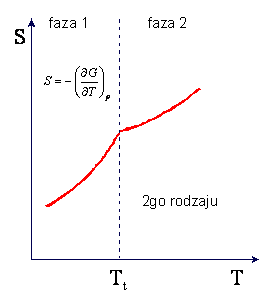
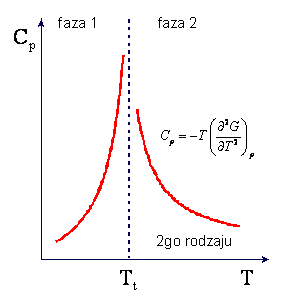
Kształt krzywej ciepła właściwego
Cp przypomina grecką literę λ i z tego powodu przemiana II-go
rodzaju jest czesto nazywana punktem lambda.

Termin
„zjawiska transportu” jest określeniem zbiorczym dla szerokiej
klasy przepływów. Dotyczy to zwłaszcza przepływu ładunku elektrycznego (prądu),
ciepła, masy (dyfuzja) i pędu (lepkość). Zjawiska transportu występują tylko w
stanach nierównowagowych. Kiedy układ osiąga stan równowagi wtedy ustają w nim
wszelkie przepływy i przenoszenie np. ładunku i masy odbywa się tylko w wyniku
ruchów termicznych i jest tak samo szybkie „w lewo” jak i „w
prawo” nie prowadząc do żadnych zmian sumarycznych. Motorem napędzającym
każdy transport jest wystąpienie spadków (gradientów) odpowiednich wielkości. Oznacza
to, że jeżeli w pewnym miejscu wartość interesującej nas wielkości wynosi Z1
to w punkcie oddalonym o Δx ta wartość spada do Z2. Jeżeli ten
spadek, ΔZ = Z2 – Z1, odbywa się liniowo to
stosunek ΔZ/Δx jest równy dokładnie gradientowi wielkości Z, grad Z.
Jeżeli ten spadek odbywa się nieliniowo (w dowolny sposób), to gradient
określamy jako stosunek odpowiednich różniczek, grad Z = dZ/dx. Ponieważ
transport odbywa się w kierunku spadku wielkości Z to ten kierunek jest zgodny
z ujemną wartością gradientu, czyli zgodny z „- grad Z”.

Przedstawiony
poniżej skrótowy opis poszczególnych zjawisk transportu jest wystarczająco jednoznaczny,
ponieważ zastosowano tutaj standardowe oznaczenia wszystkich występujących
wielkości.
Transport
ładunku (prąd elektryczny)
![]()
![]()
σ jest przewodnością ośrodka.
Transport ciepła
![]()
λ jest współczynnikiem przewodnictwa cieplnego.
Transport
masy (dyfuzja)
![]()
c - koncentracja, D - współczynnik dyfuzji.
Transport
pędu (lepkość)
![]()
η - współczynnik lepkości.