GAZY RZECZYWISTE
Równanie
van der Waalsa. Przemiany fazowe I-go i II-go rodzaju. Reguła faz Gibbsa.
* Niektóre inne równania stanu
* Przemiany fazowe I-go i II-go rodzaju

Gazy rzeczywiste nie mają już ograniczeń nakładanych na
gazy idealne (którymi były brak oddziaływań między cząstkami i brak rozmiarów
cząstek).
Ponieważ między cząstkami rzeczywistymi mogą występować różnorodne i często krańcowo skomplikowane oddziaływania wzajemne, a kształt molekuł jest często bardzo złożony, to nie jest możliwe stworzenie jednego uniwersalnego i ścisłego równania stanu. Jak pamiętamy, takie równanie stanu istnieje i ma ono postać
pV = RT
ale obowiązuje ono dla gazu idealnego, który naprawdę nie
istnieje. - Nie znamy, niestety, ścisłego równania stanu dla któregokolwiek
z gazów rzeczywistych.

Przybliżonym równaniem stanu, które uwzględnia istotne
dla gazu rzeczywistego czynniki: oddziaływanie na odległość istniejące między
cząstkami (atomami, molekułami) gazu rzeczywistego i ich rozmiary własne jest
równanie van der Waalsa:
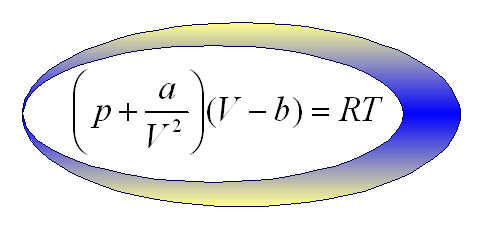
Oddziaływanie między cząstkami jest zawsze przyciągające i gaz rzeczywisty zachowuje
się tak, jak gdyby na niego działało dodatkowe ciśnienie a/V2, a stała
b uwzględnia istnienie objętości własnej molekuł, niedostępnej już dla innych
molekuł znajdujących się w tej samej objętości całkowitej V.
Stałe a i b w równaniu van der Waalsa
![]()
są dobierane oddzielnie dla każdego konkretnego gazu. Zaletami
równania van der Waalsa są jego prostota, oraz fakt, że przewiduje ono wartości
krytyczne i to, że można w nim określić odcinki skraplania gazu.
Gaz, w którym
cząstki przyciągają się wzajemnie, skupia się bardziej niż wynikało by to z
ciśnienia zewnętrznego p i dlatego opisując gaz należy dodać do ciśnienia p
dodatkową wartość wynoszącą w tym równaniu a/V2.
Gaz rzeczywisty zachowuje się zatem tak, jak gdyby na
niego działało dodatkowe ciśnienie. Jednak ciśnienie gazu mierzone na zewnątrz
będzie, oczywiście, mniejsze, jak to można pokazać symbolicznie na rysunku.
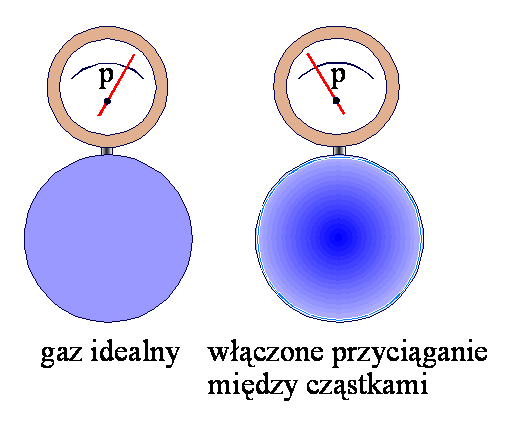
Ciśnienie gazu mierzone na manometrze będzie, oczywiście, mniejsze w przypadku gazu rzeczywistego, podczas gdy w równaniu van der Waalsa przewidujemy ciśnienie większe o czynnik a/V2. Postępujemy tak, dlatego, że naszym zadaniem jest opisać zachowanie się gazu a nie manometru.
Izotermy van der Waalsa, czyli wykresy p = p(V) dla
temperatur T1 < T2 < T3 .... otrzymane
na podstawie równania van der Waalsa mają kształt esowaty.
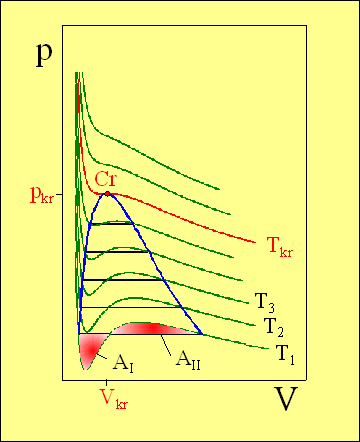
Jest to kształt silnie „niefizyczny” ponieważ
przewiduje na pewnych odcinkach zmniejszanie się ciśnienia wraz ze
zmniejszaniem się objętości. Okazuje się jednak, że na tych izotermach można
poprowadzić odcinki poziome, które obrazują proces skraplania / parowania i
odwzorowują prawdziwe, obserwowane eksperymentalnie zachowanie się gazu
rzeczywistego. Pola powierzchni AI i AII są dla
określonej izotermy jednakowe. Punkt krytyczny Cr znajduje się na izotermie
krytycznej Tkr i matematycznie jest dla tej izotermy punktem
przegięcia.
Widać, że
gazu nie można skroplić – niezależnie od przyłożonego ciśnienia –
jeżeli jego temperatury nie obniżymy poniżej temperatury krytycznej.
Ponieważ punkt krytyczny Cr znajdujący
się na izotermie krytycznej Tkr
jest dla tej krzywej punktem przegięcia to oznacza to, że w punkcie
krytycznym
![]() oraz
oraz 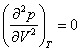
Z tych równań oraz z samego równania van der Waalsa znajdujemy
parametry krytyczne Tc, pc i Vc w postaci
![]()
Współczynnik krytyczny
dla równania stanu van der Waalsa wynosi
![]()
Wartości znalezione eksperymentalnie wahają się od 3.03 dla
H2 do 4 dla dużych molekuł. Stosując parametry zredukowane
![]()
otrzymujemy równanie stanu nazywane zredukowanym równaniem
stanu
![]()
|
Niektóre inne równania stanu Pewne równania stanu, inne niż
równanie van der Waalsa, znalazły szczególne zastosowania i w pewnych
przypadkach dają lepszą zgodność z eksperymentem w ograniczonych zakresach
parametrów. Jednakże są one nieodmiennie trudniejsze do matematycznych opracowań.
Równanie Dieterici
przewiduje istnienie punktu
krytycznego z Równanie Berthelota
w którym stała a’ jest niezależna od temperatury. Równanie to jest szczególnie wygodne w zastosowaniach do gazów i par w niskich temperaturach. Równanie Callendara wprowadzone wyłącznie dla wody i nie
przewidujące punktu krytycznego
|

Przemiany fazowe są szczególnego rodzaju procesami
termodynamicznymi, w których zmiana stanu układu jest połączona ze zmianą fazy
substancji tworzącej ten układ. Proces skraplania gazu rzeczywistego, o którym
mówiliśmy przy okazji równania van der Waalsa, jest przykładem przemiany
fazowej chociaż samo równanie van der Waalsa nie przewiduje skraplania i
odcinki izoterm odpowiadające skraplaniu wprowadziliśmy na podstawie danych
eksperymentalnych.
Wszystkie, dobrze znane z życia codziennego zmiany stanu
skupienia, jak
parowanie
skraplanie
sublimacja
osadzanie
topnienie
krystalizacja
noszą nazwę przemian fazowych
I-go rodzaju.
Przemiana fazowa I-go rodzaju
zachodzi wtedy, gdy w tym procesie obserwuje się:
-
Niezerowe ukryte ciepło (entalpia) przemiany;
-
Występowanie nukleacji (zarodnikowania nowej fazy), czyli że przemiana nie zachodzi
w całej objętości jednocześnie;
- Istnienie zjawisk “prze” (przechłodzenia i przegrzania).
Najlepiej znanymi przykłada przemian fazowych I-go
rodzaju są powszechnie znane zmiany stanu skupienia : parowanie,
skraplanie, topnienie, krystalizacja.
Zjawiska przemian fazowych są rezultatem powszechnej w przyrodzie tendencji każdego układu do osiągnięcia stanu, w którym energia tego układu jest najmniejsza.
Wygodnym do analizy przemian fazowych rodzajem energii jest tzw. energia swobodna F = U - TS, czyli energia, która w całości może zostać zamieniona na pracę. Entropia S zostanie omówiona później w kontekście II. zasady termodynamiki.
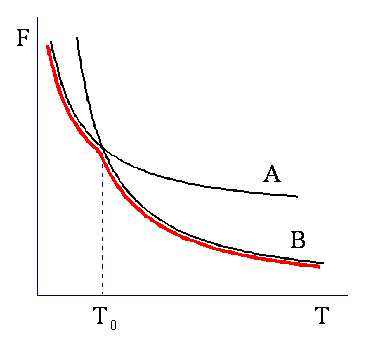
Wykresy energii F w funkcji temperatury dla dwóch stanów układu A i B są przedstawione czarnymi liniami. W niższej temperaturze stabilny – wygodniejszy energetycznie - jest stan A (ma niższe F), a w wyższej stabilny jest B (teraz on ma niższe F). W czasie ogrzewania układ przesuwa się po czerwonej linii.
W temperaturze
T0 zachodzi przemiana fazowa ze stanu A do B. W tym punkcie energie
swobodne obu faz są jednakowe: FA = FB. W całym procesie
ogrzewania układ przyjmuje najniższą dopuszczalną dla niego wartość energii
swobodnej.
Fazy, jakie może przyjmować układ, wygodnie jest przedstawiać na diagramach fazowych w układzie temperatura – ciśnienie p-T:
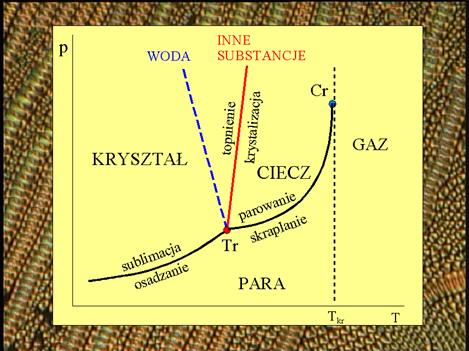
W punkcie krytycznym Cr zanika różnica między gazem a cieczą. Proszę zwrócić uwagę na odmienne nachylenie linii topnienie – krystalizacja dla wody.
Zupełnie inny charakter mają przemiany fazowe II-go rodzaju. Nie mają one żadnej z cech przemian I-go rodzaju.
W przemianie fazowej II-go
rodzaju NIE obserwuje się:
-
Niezerowego ukrytego ciepła (entalpii) przemiany;
- Nukleacji
(zarodnikowania nowej fazy), czyli że przemiana zachodzi w całej objętości
jednocześnie;
- Zjawisk “prze” (przechłodzenia i przegrzania).
W temperaturze, w której zachodzi przemiana fazowa II.
rodzaju nie ma zmiany stanu skupienia, a
występuje
tylko zmiana symetrii układu. Przykładami przemian fazowych II-go
rodzaju są:
Przemiana typu
porządek-nieporządek w stopach
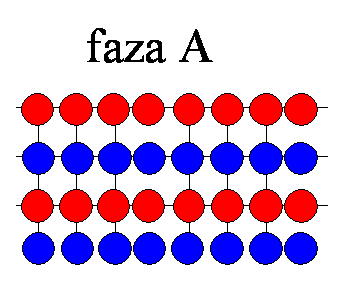
W niskiej temperaturze atomy obu rodzajów rozłożone są w oddzielnych warstwach („porządek”)
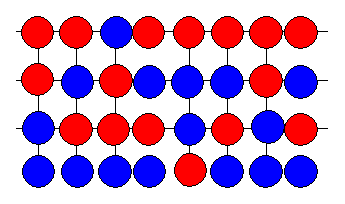
W wyższej temperaturze ten porządek ulega zaburzeniu – niektóre atomy czerwone i niebieskie wymieniają się swoimi położeniami w sieci chociaż ciągle prawdopodobieństwa ich znajdowania w poszczególnych warstwach nie są jednakowe. Jest to zaburzona faza A.
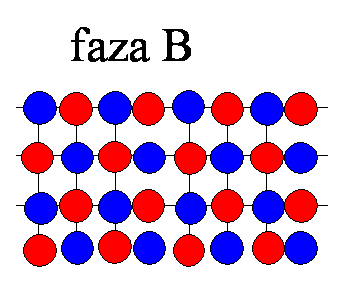
W jeszcze wyższej temperaturze oba rodzaje atomów zostają całkowicie wymieszane („nieporządek”). Prawdopodobieństwa znalezienia w określonym węźle atomu czerwonego i niebieskiego są jednakowe. Jest to faza B.
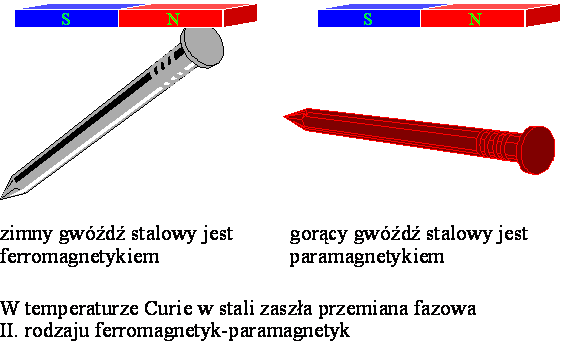
Przemiana
ferromagnetyk ↔ paramagnetyk
Bardzo gorący gwóźdź stalowy nie jest przyciągany przez magnes
Przemiana przewodnik
↔ nadprzewodnik
Prąd elektryczny raz wzbudzony w pierścieniu nadprzewodzącym będzie w nim płynął praktycznie bez końca. O płynącym prądzie świadczy wytwarzane przez niego pole magnetyczne B.
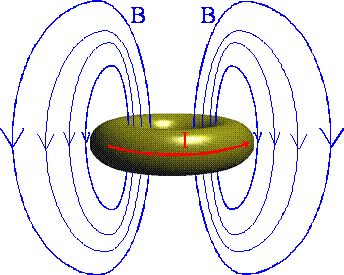
Pole magnetyczne nie wnika w nadprzewodnik, co powoduje lewitację nad nim magnesu (efekt Meissnera).
Przemiana ciecz lepka ↔ ciecz nadciekła.
W bardzo niskich temperaturach, 1 – 2 K i niższych w okolicy zera bezwzględnego, niektóre ciecze, jak hel, przechodzą w stan nadciekły.

Jednym z wielu zjawisk obserwowanych w cieczach nadciekłych jest samorzutne wypełzanie tych cieczy ze zbiorników.

Cenną wskazówką na temat zależności między liczbą
składników w układzie C (components), liczbą faz współistniejących w danym
stanie P (phases), a liczbą stopni swobody F (degrees of freedom) jest reguła
faz Gibbsa. Reguła ta jest przydatna przy przewidywaniu zachowania się układów
bardziej złożonych:
F + P = C + 2
F - liczba stopni swobody, oznacza ilość parametrów (p,
V, T, ...), które mogą być zmienione bez miany stanu równowagi układu,
P - liczba współistniejących w układzie faz, czyli tych
części, które są odseparowane od innych granicą rozdziału faz (np. roztwór wody
i soli jest jedną fazą ale nadmiar soli wraz z graniczącym roztworem stanowią
dwie fazy),
C - liczba składników (rodzajów istniejących związków chemicznych) w układzie.
Może być interesujące, że bardzo podobną postać ma słynny
wzór Eulera-Kartezjusza dla wielościanów
V + F = E + 2
V jest liczbą wierzchołków wielościanu,
F - liczbą jego ścian,
E - liczbą jego krawędzi